
All initial protein structure prediction algorithms use large language models to derive three dimensional structure prediction for a given amino acid sequence. This process is related to those employed by ChatGPT and other AI engines that also use large language models to predict an answer to questions submitted but in this case, amino acids are instead of words. For images, however a different class of algorithms is used that is based on Diffusion models. The Backer Lab, of Rosetta fame, has introduced a tool called RFDiffusion that has been trained to invent proteins – well – out of thin air.
Diffusion AI models: From noise to image.
In very simplified terms: Diffusion algorithms have been trained to learn how to undo the excessive noise added to an image of which they know what is depicted. Traditionally, un-blurring, or denoising a picture has been very challenging and only of very limited success. Using this training, however, diffusion learned how to generate images with known motif from noise. To this end, a large amount of images with known motifs have been blurred extensively (or noised) excessively and used to train diffusion AI algorithms to learn how to undo this blurring. As a result, the algorithms how to generate a picture of a given motif from a random noise input (seed). Changing the seed will generate a different output despite using the same target motif and model.
RFDiffusion
Adopting this approach for proteins, the Backer Lab has trained the RFDiffusion algorithm on all know protein structures in order to give it the ability to it to generate (or invent) new proteins from a cloud of randomly positioned amino acids in a 3D space.
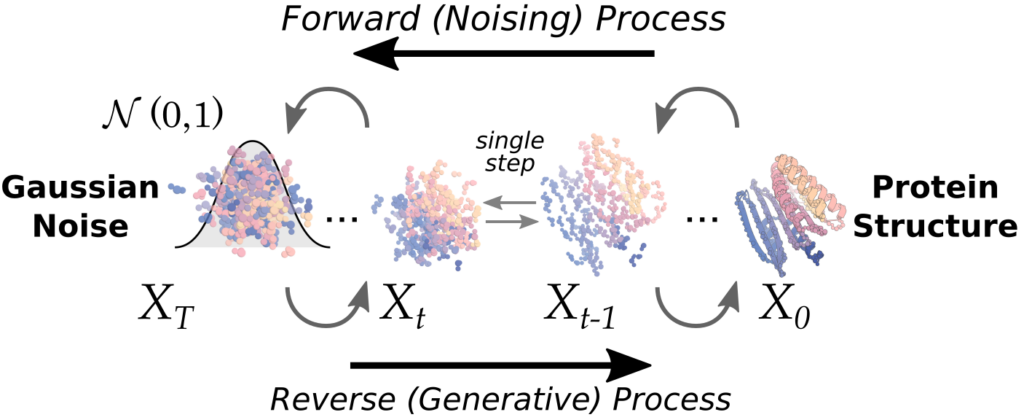
These algorithms, however, typically only produce backbone coordinates but can be used to scaffold protein motifs, design binders, redesign protein domains and to generate multimers. However, as they do not integrate any side chain information, additional algorithms are required to come up with an amino acid sequence that will actually fold into this backbone structure.
With the release of Alphafold 3, the most successful protein structure prediction algorithm also switched from a large language model to a diffusion model, thereby reducing the algorithm’s reliance on multi-sequence alignments and therefore using considerably less computational resources for structure predictions.
From backbone structure to sequence: ProteinMPNN
Now that we have beautiful backbone coordinates for our desired proteins, we need to come up with an amino acid sequence that will fold into those backbone structures. To this end a different algorithm has been trained on all available protein structures to lear what sequences will fold into what structures. The algorithm most commonly used is ProteinMPNN. Again, depending on the seed, the algorithm will provide us with different amino acid sequences predicted to fold into this particular backbone structure. Moreover, ProteinMPNN does not integrate at side chains or protein ligands.
LigandMPNN
ProteinMPNN has been superseded by LigandMPNN, an algorithm that now also integrates side-chains, heteroatoms and non-protein Ligands. As ProteinMPNN, this algorithm is now also used to optimize proteins by letting the alogithm come up with improved sequences that will fold into a desired structure.