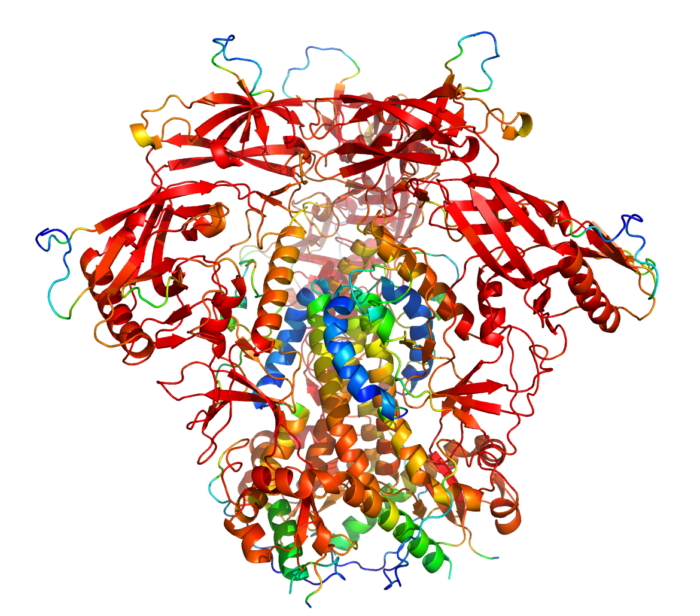
With the release of version 2 of Google’s Deepmind Alphafold software, in silico protein structure prediction has become more accurate than ever. Moreover, the software has gained the ability to predict multimer structures which is key for many immunological and virological applications. We took the past few weeks to explore the abilities and limits of this amazing piece of software. Here is what we found.
How does it work
Simply put, it uses sequence databases to form multi-sequence alignments (MSA) that it will then use to identify residues that are likely to be interacting with each other, based on the principle that evolutionary changes in amino acid positions are often accompanied with changes in other positions, indicating that the two amino acids are interacting.
The first neural block engine (Envformer, 48 blocks) uses the sequence data to define the relationship between the different AAs. This information is the used in the second module, the structure module.
The structure modules predicts a rotation and translation for each residue to place them individually in the 3-dimensional space. It then adds the side chains and forms the actual protein backbone. These prediction are then further refined by using an AMBER relaxation to remove clashes and other violations.
VRAM is the limit
The most crucial hardware requirement for running AlphaFold is the GPU. Alphafold relies on NVIDIA’s CUDA framework and will not work with other GPUs. It also requires a lot of GPU memory as memory usage (and requirement) increases quadratically to the number of residues to be folded. It therefore probably will not be able to do much on a desktop computer containing a consumer-grade GPU offering from NVIDIA. Although linking two GPUs with NVIDIA NVLink Bridges can increase the available memory, the only one GPU will be used for the number crunching.
For our tests, we have used hardware using dual NVLinked RTXA5000 and RTXA6000 GPUs with 48 and 96 GB of combined memory.
How accurate are the predictions
In order to test accuracy of Alphafold predictions, we compared the best ranked prediction for HIV envelope trimers with experimental data (PDB 5V8L). The prediction was very accurate, with an RMSD value of 1.092. Moreover, the AI algorithm also made low-confidence predictions for areas of the protein, that typically do not resolve in X-ray or cryoEM structures.
How strongly is it biased by structures that have already been published in structure databases?
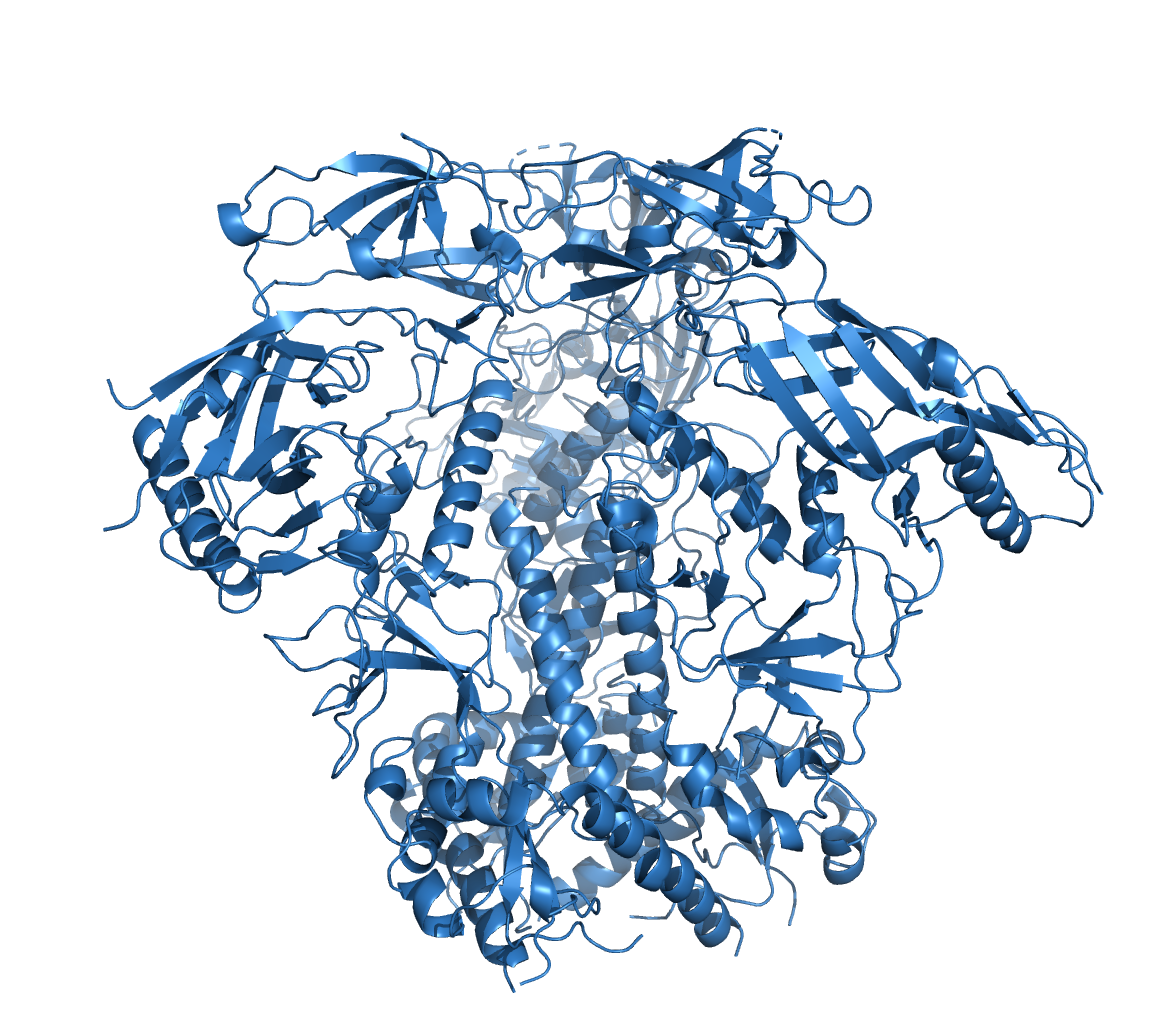
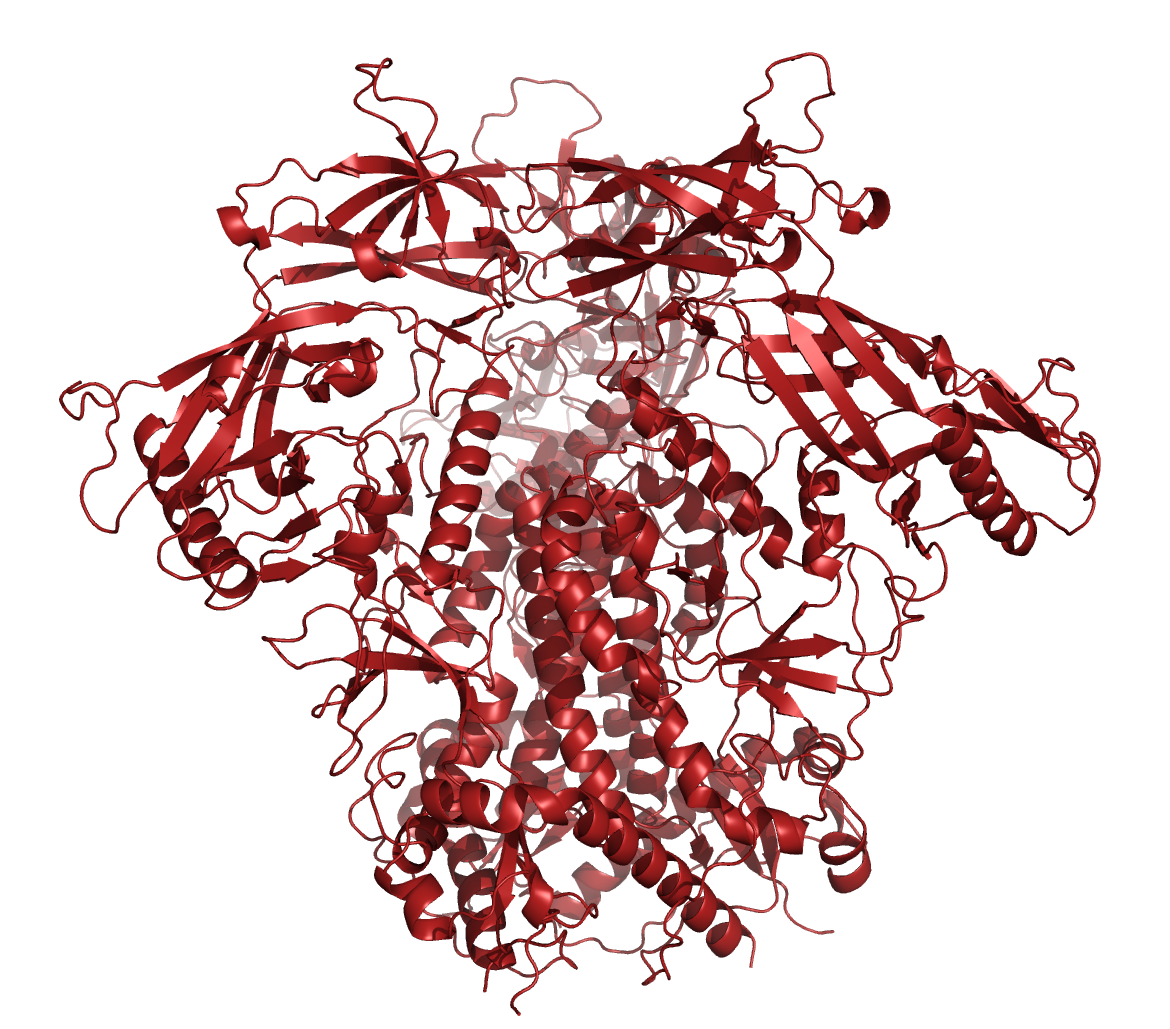
Besides the MSAs, AlphaFold can also make use of templates, i.e. related proteins whose structure have already been solved. However, AlphaFold algorithm can produce reliable structure predictions w/o templates when the MSA is large enough (i.e. hundreds of sequences). In these cases, Alphafold tends to ignore the templates altogether. Moreover, according to a blog by a DeepMind engineer, AlphaFold has a good mechanism to prevent the use of bad templates, or parts of a template that it considers misleading.
To test the impact of structure database, we ran structure predictions for the HIV BG505 envelope trimer with and without assistance of the structural databases.
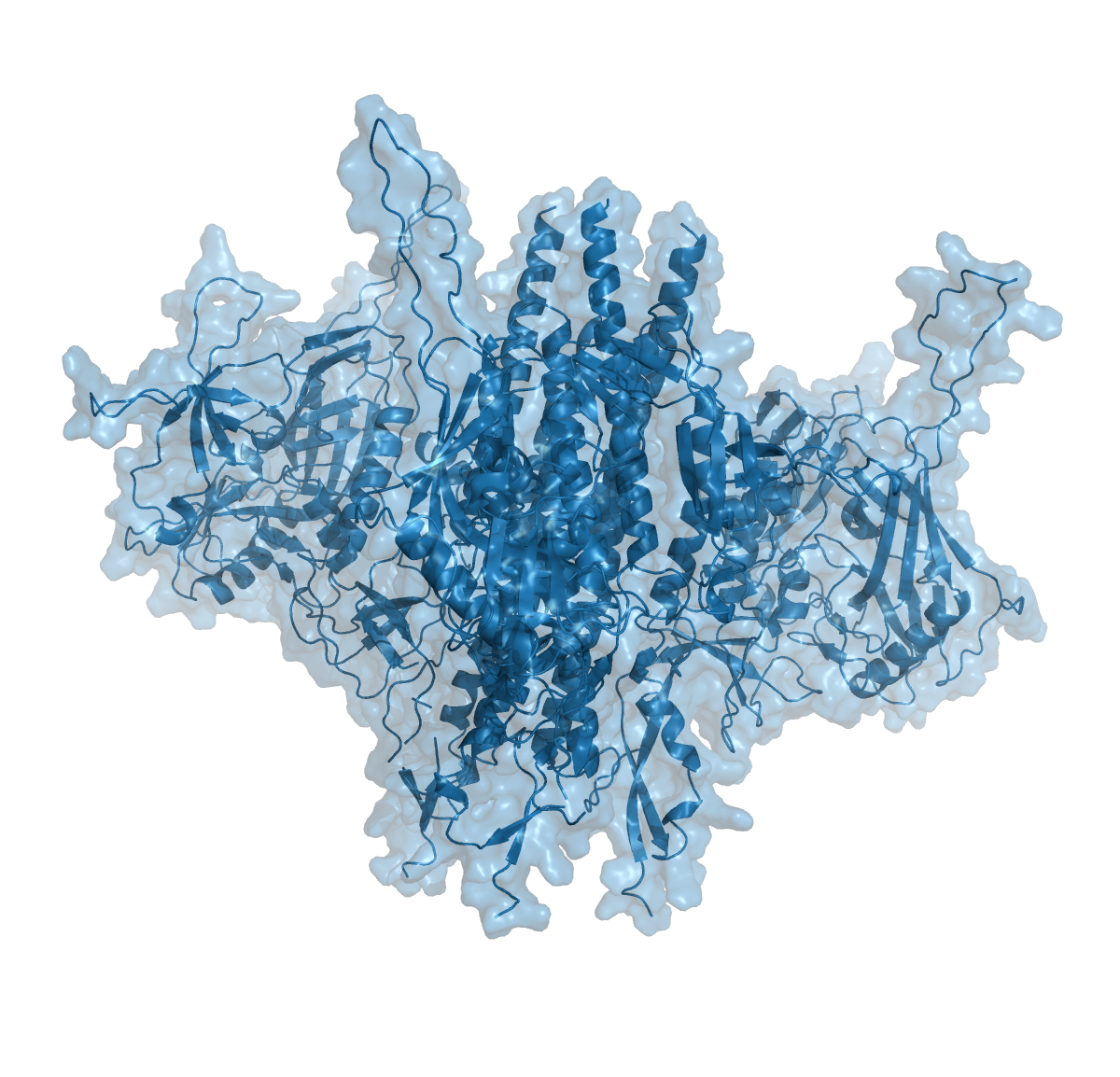
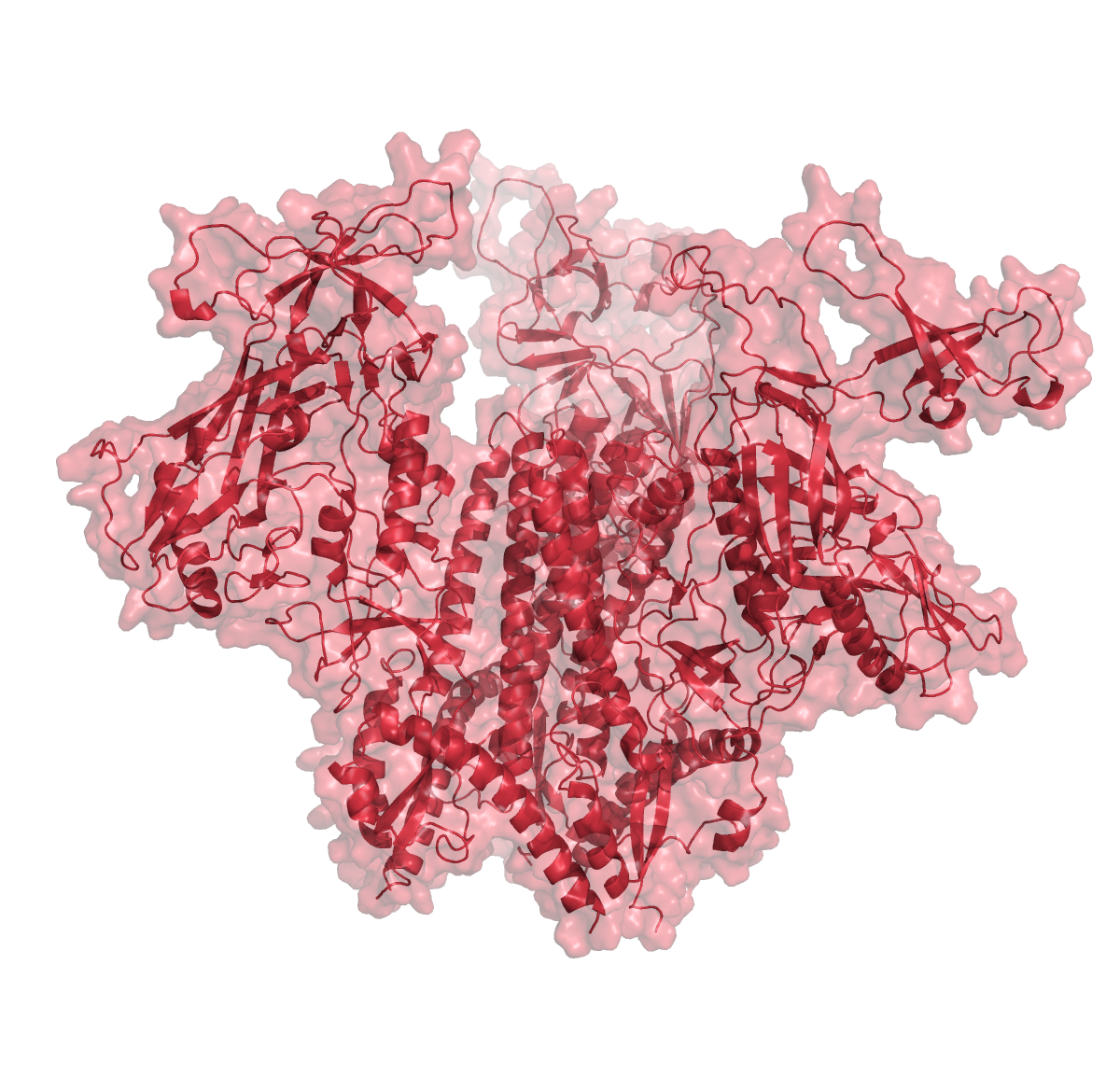

As illustrated in the figure above, the AlphaFold with PDB templates available accurately predicted the trimer structure. However, wenn the same fold was performed without PDB-templates, parts of the proteins were predicted correctly but their relative orientation was off. Interestingly, however, when stabilized versions of the HIV BG505 were folded without PDB templates, quality of the predictions improved, indicating that the algorithm recognized the stabilizing impact of the those sequence modifications. However, prediction of the spring-loaded top parts where the hypervariable loops are located, was still inaccurate.
How to interpret the predictions
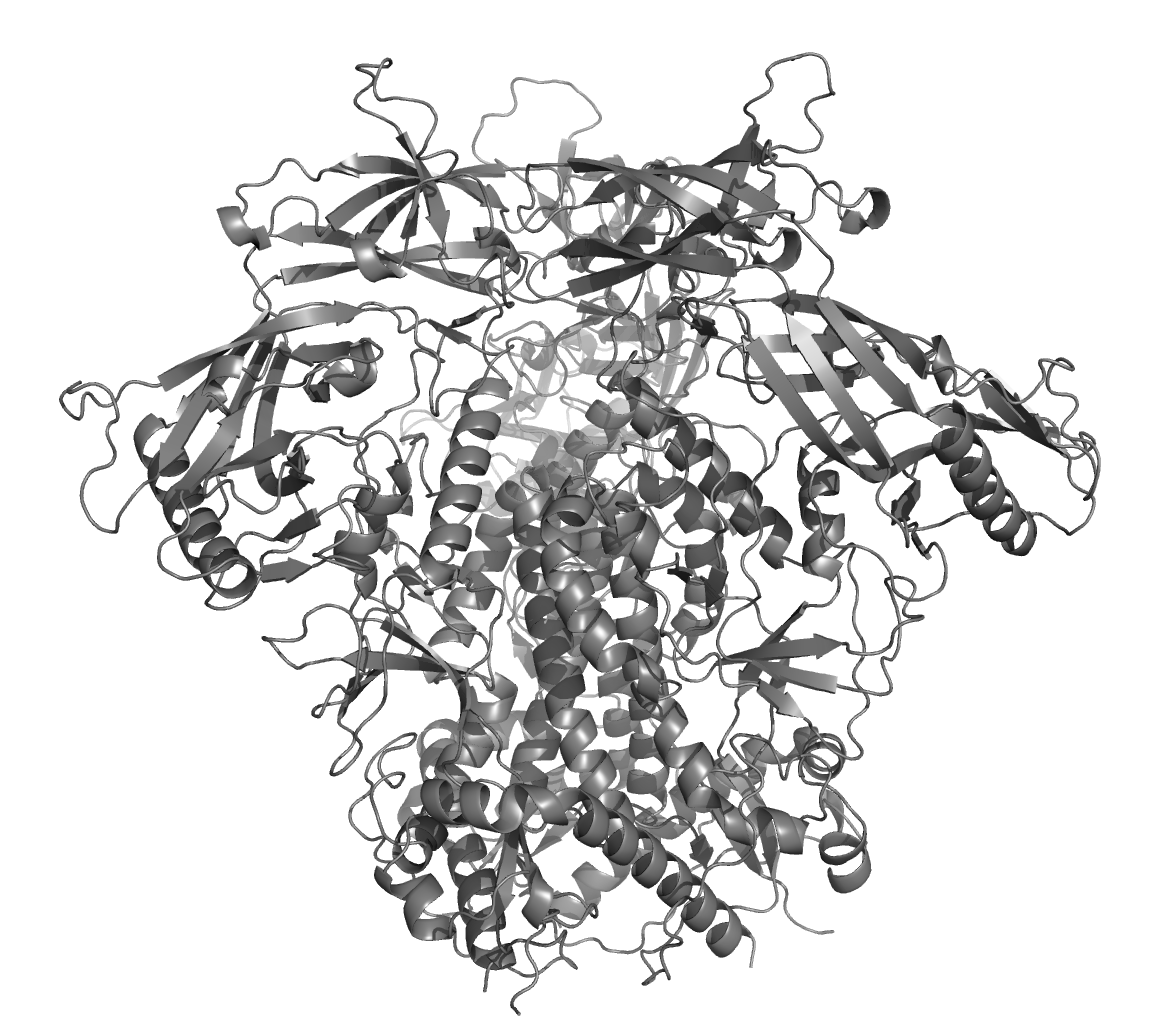
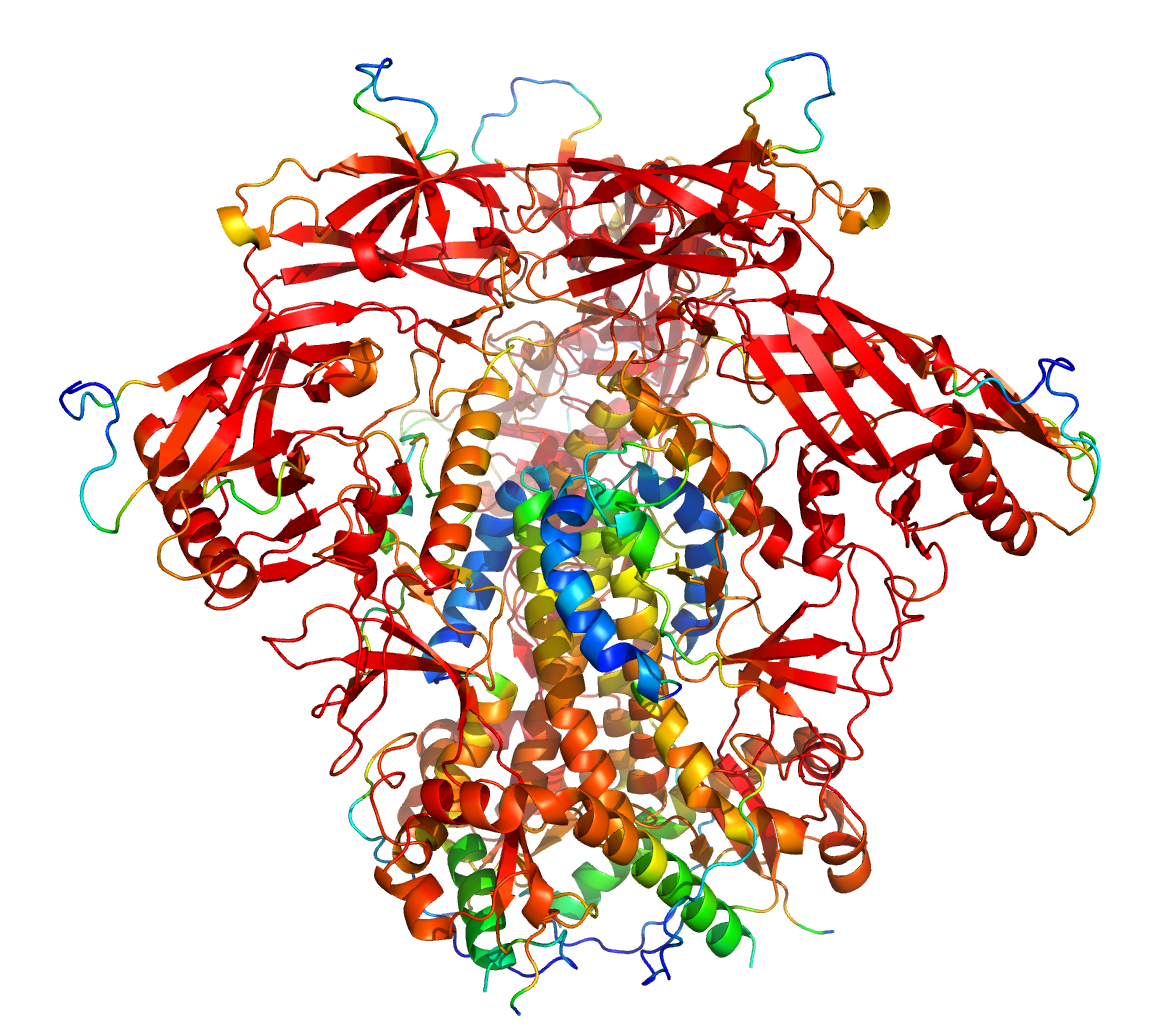
Local confidence: pLDDT
pLDDT values, ranging from 0 to 100, give an idea as to who confident alphaFold is about the local accuracy of a structure within a domain. The pLDDT value, however, does not provide information as to how confident AlphaFold is with regard to their relative domain positions.
pLDDT > 70 confident prediction,
pLDDT < 50 possibly disordered
The pLDDT values are stored as B-values in the respective PDBs from the structure predictions and can be made visible as color gradient in the most PDB viewers.
Can AlphaFold be used for antibody/epitope predictions?
We ran a number of known antibody-antigen interactions through AF2 and found that with a few exceptions, AF2 failed to predict the antibody binding, and CDR conformation for that matter, accurately. This is not surprising regarding the design and training of the software. However, we have successfully used AF2 to assist and accelerate protein designs tasks albeit the fact that AF2 has not been designed for this purpose.
Conclusions
AF2 and other AI-assisted protein structure prediction are not a magic bullet. However, with the right knowledge, AF2 can drastically accelerate protein design projects and in our experience, AI-assisted designs can often be achieved in two iterations involving a limited number of constructs.
We would be more than happy to consult and assist you with your protein design projects. We can also help you with setting up standalone and containerized versions AF2 on your own hardware.
If you have any questions, please contact us via our contact form.
Lars Hangartner, Ph.D.